
Contenuto
- Le cause
- Sintomi
- Esami e test
- Trattamento
- Gruppi di supporto
- Outlook (prognosi)
- Quando contattare un professionista medico
- Prevenzione
- Nomi alternativi
- immagini
- Riferimenti
- Data di revisione 10/15/2018
La malattia di Niemann-Pick (NPD) è un gruppo di malattie trasmesse attraverso le famiglie (ereditate) in cui le sostanze grasse chiamate lipidi si raccolgono nelle cellule della milza, del fegato e del cervello.
Esistono tre forme comuni di malattia:
- Digitare un
- Tipo B
- Tipo C
Ogni tipo coinvolge diversi organi. Può o non può coinvolgere il sistema nervoso e la respirazione. Ognuno può causare sintomi diversi e possono verificarsi in momenti diversi durante la vita.
Le cause
I tipi di NPD A e B si verificano quando le cellule del corpo non hanno un enzima chiamato sfingomielinasi acida (ASM). Questa sostanza aiuta ad abbattere (metabolizzare) una sostanza grassa chiamata sfingomielina, che si trova in ogni cellula del corpo.
Se l'ASM è mancante o non funziona correttamente, la sfingomielina si accumula all'interno delle cellule. Questo uccide le tue cellule e rende difficile il corretto funzionamento degli organi.
Il tipo A si verifica in tutte le razze ed etnie. È più comune nella popolazione ebraica ashkenazita (dell'Europa orientale).
Il tipo C si verifica quando il corpo non riesce ad abbattere correttamente il colesterolo e altri grassi (lipidi). Questo porta a troppi livelli di colesterolo nel fegato e nella milza e troppi altri lipidi nel cervello. Il tipo C è più comune tra i portoricani di origine spagnola.
Il tipo C1 è una variante del tipo C. Coinvolge un difetto che interferisce con il modo in cui il colesterolo si muove tra le cellule cerebrali. Questo tipo è stato visto solo nella popolazione canadese francese nella contea di Yarmouth, in Nuova Scozia.
Sintomi
I sintomi variano. Altre condizioni di salute possono causare sintomi simili. Le prime fasi della malattia possono solo causare alcuni sintomi. Una persona potrebbe non avere mai tutti i sintomi.
Il tipo A di solito inizia nei primi mesi di vita. I sintomi possono includere:
- Gonfiore addominale (area del ventre) entro 3 o 6 mesi
- Macchia rosso ciliegia nella parte posteriore dell'occhio (sulla retina)
- Difficoltà di alimentazione
- Perdita delle capacità motorie iniziali (peggiora nel tempo)
I sintomi di tipo B sono generalmente più miti. Si verificano nella tarda infanzia o negli anni dell'adolescenza. Gonfiore addominale può verificarsi nei bambini piccoli. Non c'è quasi nessun coinvolgimento del cervello e del sistema nervoso, come la perdita di capacità motorie. Alcuni bambini possono avere infezioni respiratorie ripetute.
I tipi C e C1 di solito colpiscono bambini in età scolare. Tuttavia, può verificarsi in qualsiasi momento tra la prima infanzia e l'età adulta. I sintomi possono includere:
- Difficoltà nel movimento degli arti che può portare a un'andatura incerta, goffaggine, problemi di deambulazione
- Milza ingrandita
- Fegato ingrandito
- Ittero alla nascita (o poco dopo)
- Difficoltà di apprendimento e declino intellettuale
- Convulsioni
- Discorso irregolare e irregolare
- Perdita improvvisa del tono muscolare che può portare a cadute
- Tremors
- Difficoltà a muovere gli occhi su e giù
Esami e test
Un test del sangue o del midollo osseo può essere fatto per diagnosticare i tipi A e B. Il test può indicare chi ha la malattia, ma non mostra se sei un portatore. I test del DNA possono essere fatti per diagnosticare i portatori di tipi A e B.
Una biopsia cutanea viene solitamente eseguita per diagnosticare i tipi C e D. L'operatore sanitario osserva come le cellule della pelle crescono, si muovono e accumulano il colesterolo. I test del DNA possono anche essere fatti per cercare i 2 geni che causano questo tipo di malattia.
Altri test potrebbero includere:
- Aspirazione del midollo osseo
- Biopsia epatica (di solito non necessaria)
- Esame oculare con lampada a fessura
- Test per verificare il livello di ASM
Trattamento
Al momento, non esiste un trattamento efficace per il tipo A.
I trapianti di midollo osseo possono essere provati per il tipo B. I ricercatori continuano a studiare possibili trattamenti, inclusi la sostituzione enzimatica e la terapia genica.
Un nuovo farmaco chiamato miglustat è disponibile per i sintomi del sistema nervoso di tipo C.
Il colesterolo alto può essere gestito con una dieta sana o a basso contenuto di colesterolo o con medicinali. Tuttavia, la ricerca non dimostra che questi metodi impediscono il peggioramento della malattia o modificano il modo in cui le cellule abbattono il colesterolo. Le medicine sono disponibili per controllare o alleviare molti sintomi, come improvvisa perdita di tono muscolare e convulsioni.
Gruppi di supporto
Queste organizzazioni possono fornire supporto e ulteriori informazioni sulla malattia di Niemann-Pick:
- Istituto Nazionale dei Disturbi neurologici e ictus - www.ninds.nih.gov/Disorders/All-Disorders/Niemann-Pick-Disease-Information-Page
- National Niemann-Pick Disease Foundation - nnpdf.org
- Organizzazione nazionale per i disturbi rari - rarediseases.org/rare-diseases/niemann-pick-disease-type-c
Outlook (prognosi)
La NPD di tipo A è una malattia grave. Di solito porta alla morte di 2 o 3 anni.
Quelli con il tipo B possono vivere fino alla tarda infanzia o all'età adulta.
Un bambino che mostra segni di tipo C prima dell'età 1 non può vivere fino all'età scolare. Coloro che mostrano sintomi dopo essere entrati a scuola possono vivere nella loro adolescenza da metà a fine. Alcuni possono vivere nel loro 20s.
Quando contattare un professionista medico
Prendi un appuntamento con il tuo fornitore se hai una storia familiare di malattia di Niemann-Pick e hai intenzione di avere bambini. Si consiglia la consulenza genetica e lo screening.
Chiama il tuo fornitore se il tuo bambino ha sintomi di questa malattia, tra cui:
- Problemi di sviluppo
- Problemi di alimentazione
- Scarso aumento di peso
Prevenzione
Tutti i tipi di Niemann-Pick sono autosomici recessivi. Ciò significa che entrambi i genitori sono portatori. Ogni genitore ha 1 copia del gene anormale senza avere alcun segno della malattia.
Quando entrambi i genitori sono portatori, c'è il 25% di possibilità che il loro bambino abbia la malattia e il 50% di possibilità che il proprio figlio sia portatore.
Il test di rilevamento del portatore è possibile solo se viene identificato il difetto genetico. I difetti coinvolti nei tipi A e B sono stati ben studiati. Sono disponibili test del DNA per queste forme di Niemann-Pick.
Difetti genetici sono stati identificati nel DNA di molte persone con tipo C. Potrebbe essere possibile diagnosticare le persone che portano il gene anormale.
Alcuni centri offrono test per diagnosticare un bambino ancora nel grembo materno.
Nomi alternativi
NPD; Carenza di sfingomielinasi; Disturbo da accumulo lipidico - malattia di Niemann-Pick; Malattia da accumulo lisosomiale - Niemann-Pick
immagini
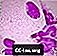
Le cellule schiumose di Niemann-Pick
Riferimenti
Kliegman RM, Stanton BF, St. Geme JW, Schor NF. Difetti nel metabolismo dei lipidi. In: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, eds. Nelson Textbook of Pediatrics. 20 ed. Philadelphia, PA: Elsevier; 2016: capitolo 86.
Patterson MC, Hendriksz CJ; Gruppo di lavoro sulle linee guida NP-C, et al. Raccomandazioni per la diagnosi e la gestione della malattia di Niemann-Pick di tipo C: un aggiornamento. Mol Genet Metab. 2012; 106 (3): 330-344. PMID: 22572546 www.ncbi.nlm.nih.gov/pubmed/22572546.
Turnpenny PD, Ellard S. Errori innati del metabolismo. In: Turnpenny PD, Ellard S, eds. Emery's Elements of Medical Genetics. 15 ed. Philadelphia, PA: Elsevier; 2017: cap. 18.
Data di revisione 10/15/2018
Aggiornato da: Anna C. Edens Hurst, MD, MS, Assistant Professor in Genetica medica, Università di Alabama a Birmingham, Birmingham, AL. Revisione fornita da VeriMed Healthcare Network. Anche recensito da David Zieve, MD, MHA, direttore medico, Brenda Conaway, direttore editoriale e A.D.A.M. Team editoriale