
Contenuto
- Le cause
- Sintomi
- Esami e test
- Trattamento
- Outlook (prognosi)
- Quando contattare un professionista medico
- Nomi alternativi
- immagini
- Riferimenti
- Data di revisione 7/26/2018
Il feocromocitoma è un raro tumore del tessuto ghiandolare surrenale. Ne risulta il rilascio di troppa epinefrina e noradrenalina, ormoni che controllano la frequenza cardiaca, il metabolismo e la pressione sanguigna.
Le cause
Il feocromocitoma può presentarsi come un singolo tumore o come più di una crescita. Di solito si sviluppa nel centro (midolla) di una o entrambe le ghiandole surrenali. Le ghiandole surrenali sono due ghiandole a forma di triangolo. Una ghiandola si trova sopra ciascun rene. In rari casi, un feocromocitoma si verifica al di fuori della ghiandola surrenale. Quando lo fa, di solito è da qualche altra parte nell'addome.
Molto pochi feocromocitomi sono cancerogeni.
I tumori possono verificarsi a qualsiasi età, ma sono più comuni dalla prima all'età adulta.
In alcuni casi, la condizione può essere vista anche tra i membri della famiglia (ereditaria).
Sintomi
La maggior parte delle persone con questo tumore ha attacchi di una serie di sintomi, che si verificano quando il tumore rilascia ormoni. Gli attacchi durano di solito da pochi minuti ad alcune ore. L'insieme dei sintomi include:
- Mal di testa
- Palpitazioni
- Sudorazione
- Alta pressione sanguigna
Man mano che il tumore cresce, gli attacchi spesso aumentano di frequenza, lunghezza e gravità.
Altri sintomi che possono verificarsi includono:
- Dolore addominale o al torace
- Irritabilità, nervosismo
- Pallore
- Perdita di peso
- Nausea e vomito
- Mancanza di respiro
- Convulsioni
- Problemi di sonno
Esami e test
Il fornitore di assistenza sanitaria effettuerà un esame fisico. Ti verrà chiesto della tua storia clinica e dei sintomi.
I test fatti possono includere:
- TAC addominale
- Biopsia surrenale
- Esame del sangue di catecolamine (catecolamine di siero)
- Test del glucosio
- Analisi del sangue metanefrina (siero metanefrina)
- Un test di imaging ha chiamato una scintigrafia MIBG
- Risonanza magnetica dell'addome
- Catecolamine di urina
- Urine metanefrine
- Scansione PET dell'addome
Trattamento
Il trattamento prevede la rimozione del tumore con un intervento chirurgico. È importante stabilizzare la pressione sanguigna e il polso con determinati farmaci prima dell'intervento chirurgico. Potrebbe essere necessario rimanere in ospedale e avere i tuoi segni vitali strettamente monitorati al momento dell'intervento. Dopo l'intervento, i tuoi segni vitali saranno monitorati continuamente in un'unità di terapia intensiva.
Quando il tumore non può essere rimosso chirurgicamente, dovrai prendere medicine per gestirlo. Di solito è necessaria una combinazione di farmaci per controllare gli effetti degli ormoni aggiuntivi. La radioterapia e la chemioterapia non sono state efficaci nel curare questo tipo di tumore.
Outlook (prognosi)
La maggior parte delle persone che hanno tumori non cancerosi che vengono rimossi con la chirurgia sono ancora vivi dopo 5 anni. I tumori tornano in alcune persone. I livelli degli ormoni norepinefrina e epinefrina tornano alla normalità dopo l'intervento chirurgico.
La pressione alta continua può verificarsi dopo l'intervento chirurgico. I trattamenti standard di solito possono controllare l'ipertensione.
Le persone che sono state trattate con successo per feocromocitoma dovrebbero sottoporsi a test di tanto in tanto per assicurarsi che il tumore non sia tornato. Anche i familiari stretti possono trarre vantaggio dai test, poiché alcuni casi vengono ereditati.
Quando contattare un professionista medico
Chiama il tuo fornitore se:
- Hanno sintomi di feocromocitoma, come mal di testa, sudorazione e palpitazioni
- Ha avuto un feocromocitoma in passato e i sintomi sono tornati
Nomi alternativi
Tumori alla cromaffina; Paraganglionoma
immagini

Ghiandole endocrine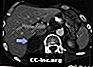
Metastasi surrenali, TAC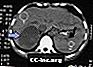
Tumore surrenale - CT
Secrezione dell'ormone ghiandola surrenale
Riferimenti
Sito web del National Cancer Institute. Trattamento con feocromocitoma e paraganglioma (PDQ) - versione per uso sanitario. Cancer.gov. www.cancer.gov/types/pheochromocytoma/hp/pheochromocytoma-treatment-pdq#link/_38_toc. Aggiornato l'8 febbraio 2018. Accesso 22 agosto 2018.
Pacak K, Timmers HJLM, Eisenhofer G. Feocromocitoma. In: Jameson JL, De Groot LJ, de Kretser DM, et al, eds. Endocrinologia: adulti e pediatrici. 7 ed. Philadelphia, PA: Elsevier Saunders; 2016: cap. 110.
Patel D, Nilubol N, Kebebew E. La gestione del feocromocitoma. In: Cameron JL, Cameron AM, eds. Terapia chirurgica corrente. 12 ed. Philadelphia, PA: Elsevier Saunders; 2017: 760-767.
Data di revisione 7/26/2018
Aggiornato da: Todd Gersten, MD, Ematologia / Oncologia, Florida Cancer Specialists & Research Institute, Wellington, FL. Revisione fornita da VeriMed Healthcare Network. Anche recensito da David Zieve, MD, MHA, direttore medico, Brenda Conaway, direttore editoriale e A.D.A.M. Team editoriale