
Contenuto
La fibrosi cistica (CF) è una malattia ereditaria pericolosa per la vita che danneggia i polmoni e il tratto digestivo. È causato da un gene difettoso che innesca la produzione di muco ispessito che ostruisce le vie respiratorie e blocca la secrezione degli enzimi digestivi.I sintomi sono progressivi e spesso gravi e possono includere problemi respiratori, infezioni polmonari ricorrenti, scarsa crescita, infertilità maschile e infiammazione cronica del pancreas, del fegato, dei reni e del cuore.
La FC può essere diagnosticata con esami del sangue, screening genetico e una procedura nota come test del cloruro di sudore.
Anche se non esiste una cura per la FC, esistono trattamenti che possono migliorare sia la durata che la qualità della vita.
Questi includono tecniche di pulizia delle vie aeree, antibiotici inalatori, diluenti del muco, enzimi pancreatici, una dieta ipercalorica e farmaci di nuova generazione noti come modulatori CFTR. Nei casi più gravi può essere necessario un trapianto di polmone.
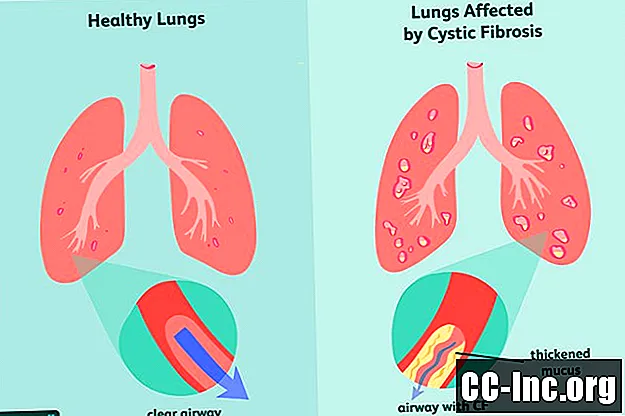
Sintomi della fibrosi cistica
In quanto malattia genetica, la fibrosi cistica è qualcosa con cui sei nato. Può o meno presentare sintomi al momento della nascita e spesso possono richiedere mesi o addirittura anni prima che compaiano segni di malattia. A quel punto, i polmoni e il tratto digestivo potrebbero aver già subito danni che non possono essere annullati.
I primi segni e sintomi più comuni della FC includono:
- Blocco delle prime feci del bambino (meconio)
- Pelle dal sapore salato
- Tosse cronica, respiro sibilante o espettorato colorato
- Feci molli, untuose e tipicamente maleodoranti
- Infezione polmonare, spesso ricorrente
- Scarsa crescita e mancanza di crescita
A meno che questi sintomi non possano essere controllati, lo stress sui polmoni (e l'incapacità di aumentare di peso) può avere un effetto cumulativo, interessando più organi e aumentando il rischio di complicanze della malattia.
Alcune delle complicazioni più caratteristiche includono:
- Pubertà ritardata
- Bronchiectasie (ispessimento cronico delle pareti polmonari)
- Perdita di peso
- Pancreatite (infiammazione del pancreas)
- Infertilità maschile
- Ipertensione polmonare (pressione sanguigna alta nei polmoni)
- Calcoli biliari
- Diabete correlato alla fibrosi cistica
- Cuore polmonare (insufficienza cardiaca destra)
- Cirrosi (cicatrici funzionali del fegato)
Poiché la FC provoca lesioni progressive alle cellule e ai tessuti, qualsiasi danno causato ai polmoni e ad altri organi sarà in gran parte irreversibile. La morte molto spesso sarà il risultato di insufficienza respiratoria, seguita da insufficienza cardiaca e insufficienza epatica.
Sintomi della fibrosi cistica
Cause
La fibrosi cistica è causata dalla mutazione del gene del recettore transmembrana della fibrosi cistica (CFTR), responsabile della produzione della proteina CFTR. Questa è la proteina di cui il corpo ha bisogno per regolare il flusso di sale e acqua dentro e fuori le cellule . Se la proteina è deformata o difettosa, può causare disidratazione sulla superficie di una cellula, portando all'ispessimento del muco circostante.
La FC è una malattia autosomica recessiva, il che significa che devi ereditare la mutazione CFTR sia da tua madre che da tuo padre per avere la malattia. Se erediti solo un gene difettoso, non avrai la FC ma sarai invece un portatore del gene mutato.
Puoi ereditare la malattia se ciascuno dei tuoi genitori ha una mutazione CFTR o la FC stessa. Se entrambi i genitori sono portatori, avresti:
- 25% di possibilità di avere la FC
- 50% di possibilità di essere un portatore
- 25 per cento di possibilità di essere inalterato
D'altra parte, se uno dei tuoi genitori è un portatore e l'altro ha la FC, hai una probabilità del 50/50 di avere la FC o di essere un portatore.
La fibrosi cistica è una delle malattie genetiche più comuni, che colpisce circa un bambino su 2.500 nati negli Stati Uniti.
È più comune tra caucasici e ispanici e si verifica meno frequentemente nelle persone di origine africana o asiatica.
Fattori di rischio della fibrosi cisticaDiagnosi
Esistono alcuni test utilizzati per diagnosticare la fibrosi cistica. Funzionano rilevando direttamente la mutazione CFTR o misurando indirettamente i cambiamenti biologici coerenti con la malattia. Il metodo di diagnosi può variare durante la gravidanza, alla nascita del bambino o in qualsiasi momento successivo.
Guida alla discussione del medico sulla fibrosi cistica
Ottieni la nostra guida stampabile per il tuo prossimo appuntamento dal medico per aiutarti a porre le domande giuste.

Dei due test standard comunemente usati per diagnosticare la FC:
- Test del cloruro di sudore, noto anche semplicemente come test del sudore, misura la quantità di cloruro sulla pelle. Poiché la FC interferisce con il trasferimento di sale da e verso le cellule, ci sarà un accumulo di sale nel sudore.
- Test genetico CFTR viene utilizzato per rilevare le mutazioni più comuni della mutazione CFTR. Sebbene siano note oltre 2.000 mutazioni CFTR che causano fibrosi cistica, le 23 incluse nel pannello standard rappresentano i sospetti più probabili.
Durante la gravidanza, il test genetico CFTR può essere utilizzato per testare fluidi ottenuti tramite amniocentesi o cellule estratte tramite prelievo dei villi coriali (CVS).
Screening neonatale è anche standardmente utilizzato per diagnosticare la FC ed è oggi prescritto in tutti i 50 stati e nel Distretto di Columbia Ciò che ciò comporta sarà diverso a seconda di dove vivi negli Stati Uniti. Se i risultati dello screening neonatale sono positivi, verrà utilizzato un test del sudore per confermare la diagnosi.
Come viene diagnosticata la fibrosi cisticaTrattamento
Anche se non esiste una cura per la fibrosi cistica, i progressi nel trattamento hanno esteso la durata della vita di coloro che convivono con la malattia.
Lo scopo del trattamento della FC è quadruplo: prevenire le infezioni, mantenere la funzione polmonare, normalizzare la digestione e rallentare la progressione della malattia.
Tra gli strumenti terapeutici utilizzati per gestire la FC:
- Tecniche di clearance delle vie aeree (ACT) vengono eseguiti per rimuovere ed espellere il muco accumulato dai polmoni. Le tecniche includono tosse sbuffata, percussioni toraciche o oscillazioni della parete toracica.
- Una dieta ricca di grassi e ipercalorica viene utilizzato per compensare il malassorbimento di grassi, proteine e sostanze nutritive nell'intestino.
- Integratori di enzimi pancreatici sono utilizzati per rafforzare gli enzimi digestivi che il pancreas non può produrre a causa dell'eccessivo accumulo di muco.
- Antibiotici vengono presi quotidianamente per prevenire infezioni polmonari batteriche.
- Mucolitici-possono essere usati farmaci usati per diluire il muco prima degli ACT.
- Modulatori CFTR sono una nuova classe di farmaci in grado di correggere alcuni difetti della proteina CFTR e ripristinarne la funzione regolatrice.
- Ossigenoterapia può essere utilizzato durante gli episodi acuti quando la respirazione è stata gravemente compromessa.
- Nutrizione enterale, noto anche come alimentazione mediante sondino, può essere utilizzato se non è possibile sostenere il peso attraverso la normale alimentazione.
- Trapianto di polmone viene considerato quando i polmoni non possono più supportare la sopravvivenza senza ventilazione meccanica.
Affrontare
Nel 1938, quando la fibrosi cistica fu classificata per la prima volta come una malattia, i bambini raramente vivevano oltre il primo anno di vita. Negli anni '80 ci si poteva aspettare di vivere dai 20 ai 25 anni. Oggi, il quadro è cambiato completamente con le persone che vivono ben oltre i 40 e persino i 50 anni se il trattamento viene iniziato presto e rispettato.
Questo non significa che la FC sia meno grave di quanto non sia mai stata. È un evento che cambia la vita, che richiede diligenza e coerenza non solo per far fronte alla malattia, ma anche per vivere il più alto standard di vita possibile.
A tal fine, è necessario normalizzare la FC nella tua vita stabilendo le routine e le pratiche per evitare gli alti e bassi che possono causare stress e aumentare la disabilità. Tra le considerazioni, dovresti:
- Gestisci la tua alimentazione. Le persone con FC spesso hanno bisogno del doppio delle calorie giornaliere rispetto alle altre persone.
- Fare esercizio regolarmente. Le routine di fitness dovrebbero idealmente prevedere un minimo di 20-30 minuti di attività aerobica tre volte a settimana. Trova qualcosa di divertente che puoi fare per tutta la vita.
- Mantieniti ben idratato. In questo modo i polmoni e l'intestino funzioneranno correttamente. A seconda della tua età, dovresti bere non meno di sei-otto bicchieri alti di acqua al giorno.
- Eseguire correttamente la liberazione delle vie aeree. Man mano che le tue esigenze di salute cambiano, anche i tipi di strumenti di autorizzazione di cui hai bisogno possono cambiare. Parla con il tuo pneumologo o fisioterapista se non stai ottenendo i risultati che dovresti.
- Cerca supporto. Oltre ad amici e familiari, puoi contattare la sezione più vicina della Cystic Fibrosis Foundation (CFF) per collegarti a una rete di supporto nella tua zona.
- Cerca aiuto finanziario. Il CFF offre servizi che aiutano le famiglie a far fronte meglio agli alti costi del trattamento della FC.
Una parola da Verywell
Mentre gli screening neonatali hanno aumentato notevolmente il tasso di diagnosi di FC nei bambini, oltre il 25% delle diagnosi viene effettuato solo durante l'infanzia, l'adolescenza e la prima età adulta.
Questo è problematico perché la diagnosi e il trattamento precoci possono prevenire molte delle complicanze più gravi della FC prima che si possa fare un qualsiasi danno grave. Sebbene il trattamento non possa fermare o invertire la malattia, può garantire molti più anni senza malattia.
A tal fine, è importante conoscere i primi sintomi della FC e parlare con il medico se sospetti che tuo figlio possa avere la malattia. Ciò è particolarmente vero negli stati che eseguono lo screening solo con esami del sangue IRT, il che potrebbe portare a una diagnosi ritardata oa un risultato falso negativo fino al 5%, secondo una ricerca della University of Wisconsin School of Medicine and Public Health .
Quali sintomi ci si può aspettare dalla fibrosi cistica?