
Contenuto
- Le cause
- Sintomi
- Esami e test
- Trattamento
- Gruppi di supporto
- Outlook (prognosi)
- Possibili complicazioni
- Quando contattare un professionista medico
- Prevenzione
- Nomi alternativi
- Istruzioni per il paziente
- immagini
- Riferimenti
- Data di revisione 2/19/2018
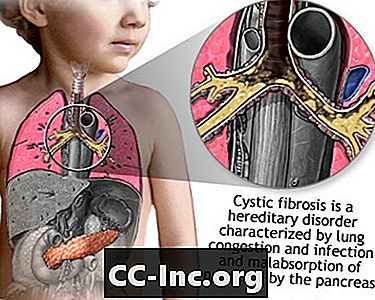
La fibrosi cistica è una malattia che causa la formazione di muco denso e appiccicoso nei polmoni, nel tratto digestivo e in altre parti del corpo. È una delle più comuni malattie polmonari croniche nei bambini e nei giovani adulti. È una malattia pericolosa per la vita.
Le cause
La fibrosi cistica (CF) è una malattia che viene trasmessa attraverso le famiglie. È causato da un gene difettoso che fa sì che il corpo produca un fluido anormalmente denso e appiccicoso, chiamato muco. Questo muco si accumula nei passaggi respiratori dei polmoni e nel pancreas.
L'accumulo di muco provoca infezioni polmonari pericolose per la vita e gravi problemi di digestione. La malattia può anche colpire le ghiandole sudoripare e il sistema riproduttivo di un uomo.
Molte persone portano un gene CF, ma non hanno sintomi. Questo perché una persona con CF deve ereditare 2 geni difettosi, 1 da ciascun genitore. Alcuni americani bianchi hanno il gene CF. È più comune tra quelli di origine nord-europea o centrale.
La maggior parte dei bambini con CF viene diagnosticata all'età di 2 anni. Per un numero ridotto, la malattia non viene rilevata fino all'età di 18 anni. Questi bambini hanno spesso una forma più lieve della malattia.
Sintomi
I sintomi nei neonati possono includere:
- Crescita ritardata
- Mancato aumento di peso normalmente durante l'infanzia
- Nessun movimento intestinale nelle prime 24 a 48 ore di vita
- Pelle dal gusto salato
I sintomi correlati alla funzione intestinale possono includere:
- Dolore alla pancia da grave stitichezza
- Aumento del gas, gonfiore o pancia che appare gonfia (distesa)
- Nausea e perdita di appetito
- Feci che sono pallide o color argilla, maleodoranti, hanno muco, o che galleggiano
- Perdita di peso
I sintomi relativi ai polmoni e ai seni possono includere:
- Tosse o aumento del muco nei seni o nei polmoni
- Fatica
- Congestione nasale causata da polipi nasali
- Episodi ripetuti di polmonite (sintomi di polmonite in qualcuno con fibrosi cistica comprendono febbre, tosse aumentata e mancanza di respiro, aumento del muco e perdita di appetito)
- Dolore o pressione del seno causati da infezione o polipi
Sintomi che potrebbero essere notati più tardi nella vita:
- Infertilità (negli uomini)
- Infiammazione ripetuta del pancreas (pancreatite)
- Sintomi respiratori
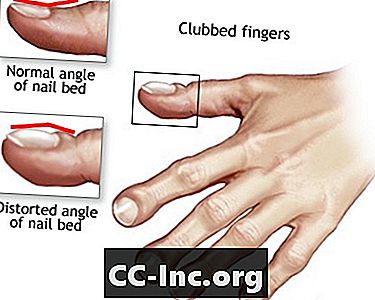
- Dita di bastoni
Esami e test
Viene eseguito un esame del sangue per aiutare a rilevare CF. Il test cerca cambiamenti nel gene CF. Altri test utilizzati per diagnosticare CF includono:
- Il test del trypsinogen (IRT) immunoreattivo è un test di screening neonatale standard per CF. Un alto livello di IRT suggerisce possibili CF e richiede ulteriori test.
- Il test del cloruro di sudore è il test diagnostico standard per la FC. Un alto livello di sale nel sudore della persona è un segno della malattia.
Altri test che identificano i problemi che possono essere correlati alla CF includono:
- Scansione a raggi X o TC del torace
- Test del grasso fecale
- Test di funzionalità polmonare
- Misura della funzione pancreatica
- Test di stimolazione della secretina
- Tripsina e chimotripsina nelle feci
- Serie di GI superiore e intestino tenue
Trattamento
Una diagnosi precoce di FC e piano di trattamento può migliorare sia la sopravvivenza che la qualità della vita. Follow-up e monitoraggio sono molto importanti. Quando possibile, si deve prestare attenzione in una clinica specializzata in fibrosi cistica. Quando i bambini raggiungono l'età adulta, devono trasferirsi in un centro specializzato in fibrosi cistica per adulti.
Il trattamento per problemi polmonari include:
- Antibiotici per prevenire e curare le infezioni del polmone e del seno. Possono essere assunti per bocca, o somministrati nelle vene o mediante trattamenti respiratori. Le persone affette da FC possono assumere antibiotici solo quando necessario o sempre. Le dosi sono spesso superiori al normale.
- Farmaci per inalazione per aiutare ad aprire le vie respiratorie.
- Altri farmaci che vengono somministrati con un trattamento respiratorio per fluidificare il muco e rendere più facile la tosse sono gli enzimi DNAse. terapia e soluzioni saline altamente concentrate (soluzione salina ipertonica).
- Vaccino antinfluenzale e vaccino polisaccaridico pneumococcico (PPV) annuo (rivolgersi al proprio medico).
- Il trapianto di polmone è un'opzione in alcuni casi.
- L'ossigenoterapia può essere necessaria in caso di peggioramento della malattia polmonare.
I problemi polmonari vengono anche trattati con terapie per assottigliare il muco. Questo rende più facile tossire il muco dai polmoni.
Questi metodi includono:
- Attività o esercizio che ti fanno respirare profondamente
- Dispositivi che vengono utilizzati durante il giorno per aiutare a liberare le vie aeree da un eccesso di muco
- Percussione del torace manuale (o fisioterapia del torace), in cui un membro della famiglia o un terapista applaude delicatamente il torace, la schiena e l'area sotto le braccia
Il trattamento per problemi intestinali e nutrizionali può includere:
- Una dieta speciale ad alto contenuto di proteine e calorie per bambini più grandi e adulti
- Enzimi pancreatici per aiutare ad assorbire grassi e proteine, che vengono assunti con ogni pasto
- Integratori vitaminici, in particolare vitamine A, D, E e K
- Il tuo fornitore può consigliare altri trattamenti se hai sgabelli molto duri
Ivacaftor e Lumacaftor sono medicinali che trattano determinati tipi di CF. Migliorano la funzione di uno dei geni difettosi che causa la CF. Di conseguenza, c'è meno accumulo di muco denso nei polmoni. Anche altri sintomi CF sono migliorati.
Cura e monitoraggio a casa dovrebbero includere:
- Evitare fumo, polvere, sporcizia, fumi, prodotti chimici domestici, fumo di camino e muffe o funghi.
- Dare fluidi abbondanti, specialmente a neonati e bambini quando fa caldo, quando ci sono diarrea o feci molli, o durante un'attività fisica extra.
- Esercitarsi 2 o 3 volte a settimana. Nuoto, jogging e ciclismo sono buone opzioni.
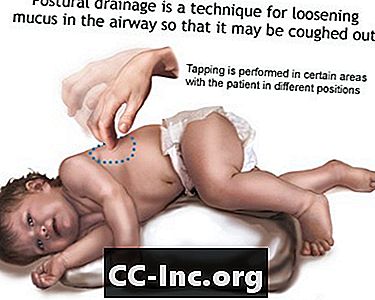
- Cancellare o allevare muco o secrezioni dalle vie respiratorie. Questo deve essere fatto da 1 a 4 volte al giorno. I pazienti, le famiglie e gli operatori sanitari devono imparare a fare le percussioni del torace e il drenaggio posturale per mantenere le vie respiratorie libere.
Gruppi di supporto
È possibile alleviare lo stress della malattia unendosi a un gruppo di supporto per la fibrosi cistica. Condividere con altri che hanno esperienze e problemi comuni può aiutare la tua famiglia a non sentirsi sola.
Outlook (prognosi)
La maggior parte dei bambini con CF rimane in buona salute fino al raggiungimento dell'età adulta. Sono in grado di prendere parte alla maggior parte delle attività e frequentare la scuola. Molti giovani adulti con CF finiscono l'università o trovano lavoro.
La malattia polmonare alla fine peggiora al punto in cui la persona è disabilitata. Oggi, la durata media della vita delle persone con CF che vivono fino all'età adulta è di circa 37 anni.
La morte è più spesso causata da complicanze polmonari.
Possibili complicazioni
La complicazione più comune è l'infezione respiratoria cronica.
Altre complicazioni includono:
- Problemi intestinali, come calcoli biliari, blocco intestinale e prolasso rettale
- Tossendo sangue
- Insufficienza respiratoria cronica
- Diabete
- Infertilità
- Malattia epatica o insufficienza epatica, pancreatite, cirrosi biliare
- Malnutrizione
- Polipi nasali e sinusite
- Osteoporosi e artrite
- Polmonite che continua a tornare
- pneumotorace
- Insufficienza cardiaca destra (cuore polmonare)
Quando contattare un professionista medico
Chiama il tuo fornitore se un bambino o un bambino ha sintomi di CF ed esperienze:
- Febbre, aumento della tosse, espettorato o espettorato di sangue nell'espettorato, perdita di appetito o altri segni di polmonite
- Aumento della perdita di peso
- Movimenti intestinali più frequenti o sgabelli maleodoranti o con più muco
- Pancia gonfia o gonfiore aumentato
Chiama il tuo fornitore se una persona con CF sviluppa nuovi sintomi o se i sintomi peggiorano, in particolare difficoltà respiratorie o tosse con sangue.
Prevenzione
La CF non può essere prevenuta. Lo screening di quelli con una storia familiare della malattia può rilevare il gene CF in molti portatori.
Nomi alternativi
CF
Istruzioni per il paziente
- Nutrizione enterale - problemi di gestione del bambino
- Tubo di alimentazione per gastrostomia - bolo
- Come respirare quando sei a corto di fiato
- Tubo di alimentazione per la giunostomia
- Drenaggio posturale
immagini
Clubbing
Drenaggio posturale
Dita di bastoni
Fibrosi cistica
Riferimenti
Accurso FJ. Fibrosi cistica. In: Goldman L, Schafer AI, eds. Medicina Goldman-Cecil. 25 ed. Philadelphia, PA: Elsevier Saunders; 2016: capitolo 89.
Egan ME, Green DM, Voynow JA. Fibrosi cistica. In: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, eds. Nelson Textbook of Pediatrics. 20 ed. Philadelphia, PA: Elsevier; 2016: cap 403.
Farrell PM, White TB, Ren CL, et al. Diagnosi della fibrosi cistica: linee guida di consenso della Fondazione per la fibrosi cistica. J Pediatr. 2017; 181S: S4-S15.e1. PMID: 28129811 www.ncbi.nlm.nih.gov/pubmed/28129811.
Rowe SM, Hoover W, Solomon GM, Sorscher EJ. Fibrosi cistica. In: Broaddus VC, Mason RJ, Ernst JD, e altri, eds. Manuale di Murray e Nadel sulla medicina respiratoria. Sesto ed. Philadelphia, PA: Elsevier Saunders; 2016: cap. 47.
Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-ivacaftor in pazienti con fibrosi cistica omozigote per phe508del. N Engl J Med. 2017; 377 (21): 2013-2023. PMID: 29099344 www.ncbi.nlm.nih.gov/pubmed/29099344.
Data di revisione 2/19/2018
Aggiornato da: Neil K. Kaneshiro, MD, MHA, Professore clinico di Pediatria, Scuola di Medicina dell'Università di Washington, Seattle, WA. Anche recensito da David Zieve, MD, MHA, direttore medico, Brenda Conaway, direttore editoriale e A.D.A.M. Team editoriale